在分子动力学Molecular Dynamics, MD)研究中,(Ab Initio Molecular Dynamics, AIMD)因能够结合量子力学计算与分子动力学模拟而成为研究复杂化学体系的重要方法。
常规AIMD模拟体系牛顿运动方程与量子力学能量面反应路径为解决这一问题,限制性(Constrained AIMD,也称受限AIMD)被提出。该方法通过在体系中对某些自由度(如键长、键角、反应坐标等)施加约束,使体系在指定的坐标空间内演化,从而强制系统探索特定的反应路径或局部能量区域。
自由能曲线电催化界面反应、溶液反应动力学、化学键断裂与形成过程DOI:10.1039/d3sc04740g
限制性AIMD的基本原理
AIMD的是。约束条件通常,例如两个原子间的距离、三原子之间的键角,或更复杂的集体变量。通过对这些自由度施加固定或可调的约束,研究者能够控制体系沿着预设路径进行演化,从而采样到原本难以获取的构型区域。
常见的约束方法Lagrange乘子法与伪势方法。例如,通过Lagrange乘子将目标反应坐标固定在某一数值上,体系的其他自由度则在约束条件下自由演化。这种方法既保证了体系的量子力学能量计算精确性,又能够有效控制模拟过程,使其聚焦于特定反应路径或构型转变。这种方法本质上是一种受控采样策略,能够为自由能面构建提供关键数据点。
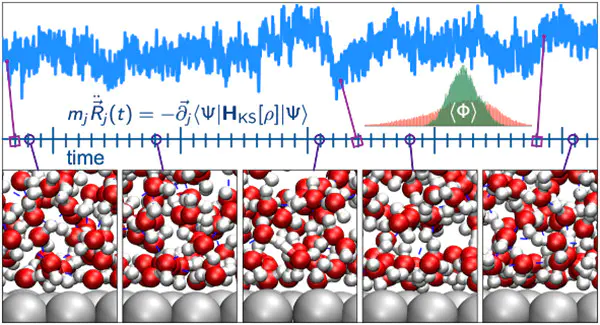
DOI:10.1021/acs.chemrev.1c00679
自由能曲线计算与反应机理解析
AIMD的最重要应用之一是(Free Energy Profile)的计算。自由能曲线是理解反应动力学与热力学本质的关键工具,它揭示了体系沿反应坐标的能量变化规律。
具体而言,研究者可以在下运行限制性这种方法被广泛应用于研究化学键断裂、质子转移、离子迁移等过程。2形成的自由能势垒,从而为理解反应速率控制步骤提供了理论依据。
DOI:10.1126/sciadv.adu1602
与过渡态搜索方法的比较与联系
AIMD在研究反应机理时,常常与过渡态搜索方法相比较。传统的过渡态搜索方法,如爬山NEB(Nudged Elastic Band),能够在0 K能量面上寻找最小能量路径(MEP)与过渡态。然而,这些方法忽略了有限温度下的热振动效应与溶剂环境效应,难以准确描述真实反应条件。
AIMD的优势在于其例如,在溶液界面反应中,水分子的氢键网络对反应路径有重要影响,常规NEB方法无法精确体现,而限制性AIMD则能在显式溶剂环境下对反应坐标进行约束与采样,得到更接近实验条件的自由能曲线。因此,DOI:10.1021/jacs.4c06629
在电催化与界面反应中的应用
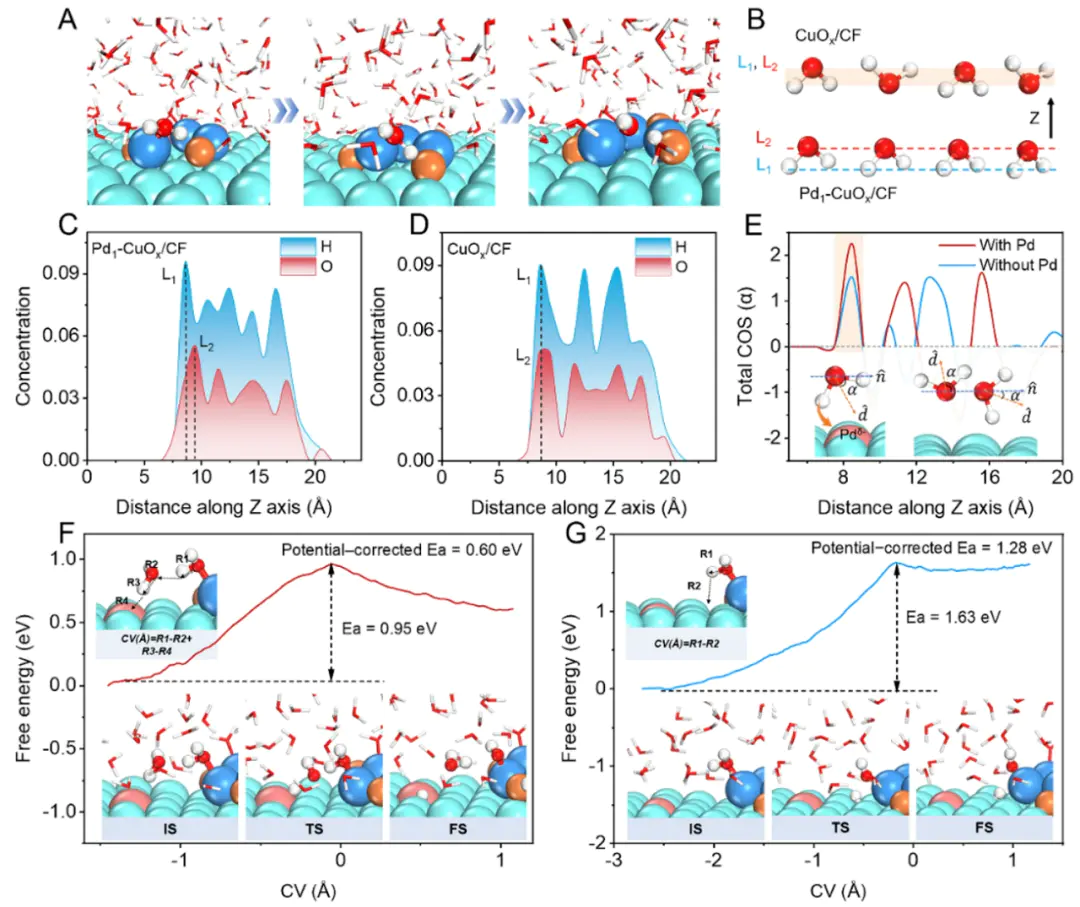
电催化研究AIMD的重要应用领域。在电极–电解质界面,反应中间体的吸附、转移与转化往往涉及复杂的溶剂效应与电荷转移过程。常规静态计算方法往往将电极与溶液分离处理,难以捕捉真实的动力学过程。而限制性AIMD能够在显式水分子、电极表面与反应物共同作用下,通过对关键反应坐标的限制,逐步描绘出完整的反应自由能曲线。
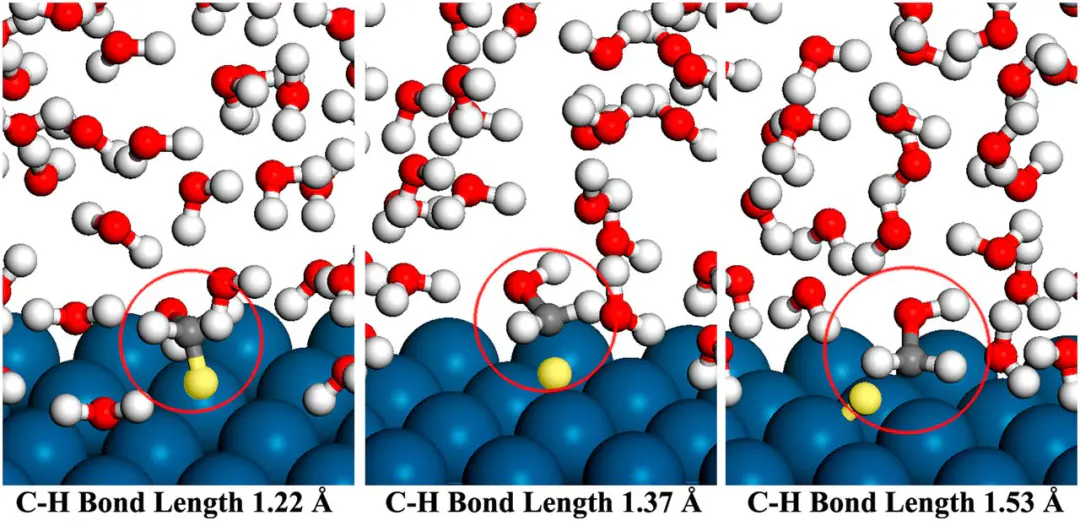
例如,在氧析出反应(2分子在电极表面逐步氢化的过程,帮助揭示反应路径与速率控制步骤。这些应用展示了限制性复杂界面电化学中的独特价值DOI:限制性最大优势局限性计算成本较高约束条件的选择对结果有重要影响此外,限制性未来,限制性其一是与增强采样方法的结合,其二是与机器学习势能面(MLP)的结合利用机器学习方法在保证量子化学精度的同时大幅降低计算成本,使得限制性AIMD能够在更大体系与更长时间尺度上应用。
AIMD已逐渐拓展到电化学储能、生物大分子反应以及材料表面工程等领域。例如,在电池研究中,限制性AIMD被用于研究锂离子在电极表面的迁移过程;在生物化学中,限制性AIMD用于探索酶催化反应中的关键步骤。这些应用表明,综上所述,AIMD是基于从头分子动力学的一种扩展方法,其通过对特定自由度施加约束,使体系能够在有限时间尺度内探索特定反应路径,从而获得自由能曲线与反应势垒。尽管其计算成本与坐标选择带来一定局限,但随着计算方法与增强采样技术的发展,限制性AIMD必将在更广阔的科学与工程领域中发挥关键作用,为揭示复杂化学与物理过程提供深刻见解。







