说明:本文华算科技介绍了界面能的基本概念、计算方法及其在材料科学中的应用,重点阐述了界面能作为表征界面原子键合环境关键参数,如何影响电荷分离与材料性能,并结合密度泛函理论(DFT)计算与实验表征,探讨通过界面工程调控界面能的设计策略。
理论指导界面能两种不同相或相同相但不同取向的晶体之间界面处单位面积的能量这个概念源于材料科学中对相界和晶界能量特性的研究,。从热力学角度来看,界面能是形成新界面所需的最小功,代表着系统自由能的增加。
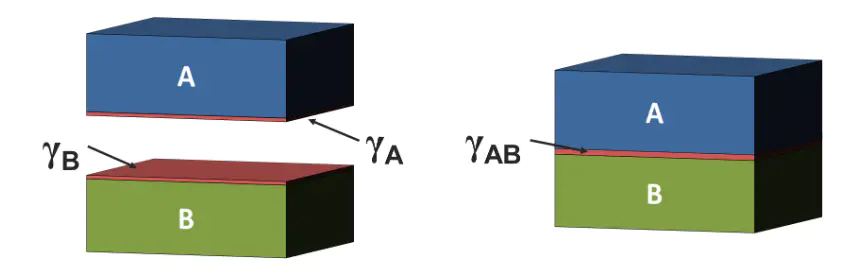
固–液界面能固–固界面能液–气界面能其中晶界能又可根据相邻晶粒的取向差分为(θ)和(θ>10°),两者的能量特征和变化规律有显著差异。
物理本质不饱和键和晶格畸变界面能的精确计算和测量对材料设计和性能预测至关重要。目前,研究者开发了多种理论计算和实验测量方法。
基于量子力学,可直接从原子层面预测界面性质。以密度泛函理论(DFT)为例,界面能可通过以下公式计算:
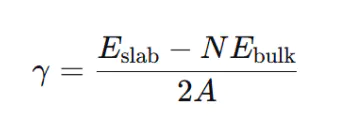
slabbulk1、案例背景与问题提出
调控界面能来增强电荷分离的创新策略DOI:10.1038/s41467-022-35502-z
电荷分离2、创新点与解决方案
构建核/壳型助催化剂结构来调整界面能BiVO4CoOxDOI:10.1038/s41467-022-35502-z
降低了BiVO面的肖特基势垒,而不影响表面反应,从而增强了界面能学的不对称性和电荷分离效率3、技术方法与表征手段
多种先进的表征技术来验证界面能调控的效果BiVO4BiVO4/AgBiVO4/PdBiVO4/(Ag/Pd)瞬态吸收光谱进一步的光载流子分布模拟显示,在BiVO4{010}DOI:10.1038/s41467-022-35502-z
这项研究证实了是增强电荷分离的一种普遍有效的方法,为设计高性能光催化剂提供了新思路。通过合理设计界面结构,在不影响表面反应的前提下优化电荷分离效率,解决了光催化领域的关键挑战。
总结
从微观的原子间相互作用到宏观的材料性能,界面能发挥着桥梁作用,连接不同尺度的现象与机制。
,界面能研究正从“表征解释“向“预测设计“转变