之前GTPCelltype的推出大大减轻了手动注释的工作量,考虑到国内用户在使用GPT的时候又很多不可避免的问题,数信院技术团队在第一时间就结合数信院AI助手专门对包进行了改动,以供大家国内使用。近期,GPTCelltype又有了新的更新,数信院肯定第一时间进行了包的升级维护,并且将包上传到了github,来,咱们来走一遍流程。
01安装与加载
首先,如果加载了GPTCelltype官方的包,我们需要取消加载并卸载原有的包:
detach("package:GPTCelltype", unload = T)detach("package:openai", unload = T)remove.packages("GPTCelltype") remove.packages("openai")
然后安装数信院AI助手的适配包,AI助手本身调用的是4o模型,因此效能相同,不必担心:
install.packages("remotes")remotes::install_github("EddieLv/openai")remotes::install_github("EddieLv/GPTCelltype")
API的获取:
首先注册数信院AI:
http://sxygptcloud.com:3000/login


淘宝下单,找客服购买一定额度,然后客服会发兑换码:

十美刀足够五个课题的多次注释:

回到之前的界面,复制令牌:
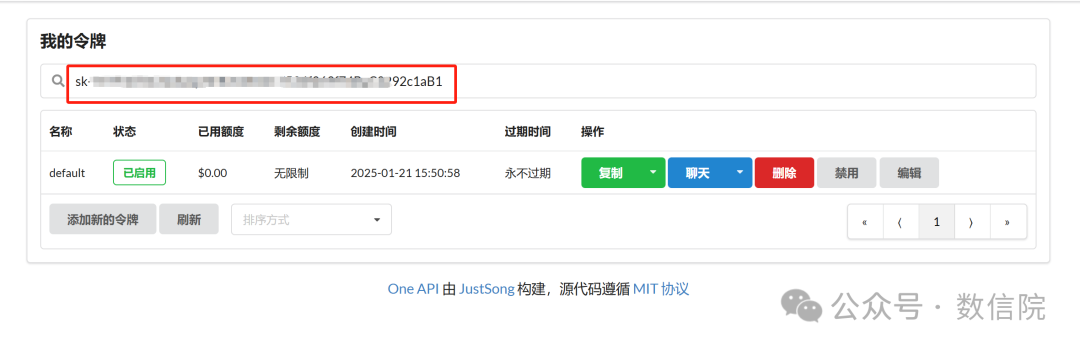
这个令牌就是后续调用注释的API码了,将在后续的步骤使用。
0设置API为环境变量:
将数信院AI助手的API码复制并添加至下方代码中
Sys.setenv(OPENAI_API_KEY = 'sk-XXXX')
0运行基本流程
library(GPTCelltype)library(openai)
GPTCelltype主要功能是 gptcelltype()。它可以通过 OpenAI GPT 模型在 Seurat 管道中或使用自定义基因列表对细胞类型进行注释。如果 gptcelltype() 用于 Seurat 管道,则需要先运行 Seurat 的 FindAllMarkers() 函数,Seurat 生成的差异基因表将作为输入。如果输入是自定义基因列表,则每个基因元素将被分配一个细胞类型。输入参数中,input 可以是 Seurat FindAllMarkers() 函数返回的差异基因表,或者是一个基因列表。tissuename是组织名称。model 是一个有效的 GPT-4 或 GPT-3.5 模型名称,列出了在 Models 页面上的模型名称,默认值为 ‘gpt-4’。topgenenumber 是当输入为 Seurat 差异基因时,用于注释的前 N 个差异基因数量。输出是一个细胞类型的向量。
library(Seurat, quietly = TRUE)
在下面的示例中,我们将使用 Seurat 包提供的一个 Seurat 对象,名为 ‘pbmc_small’。在实际应用中,应该准备一个在运行标准 Seurat 流程后获得的 Seurat 项目。该 Seurat 项目应该包含细胞聚类信息。如果尚未生成差异基因表,请使用 FindAllMarkers() 函数生成差异基因表。
data("pbmc_small")suppressWarnings({ all.markers <- FindAllMarkers(object = pbmc_small)})
得到了all.markers之后就可以直接开始注释了,一步就搞定
res <- gptcelltype(all.markers, tissuename = 'human PBMC', model = 'gpt-4')
## [1] "Note: OpenAI API key found: returning the cell type annotations."## [1] "Note: It is always recommended to check the results returned by GPT-4 in case of\n AI hallucination, before going to down-stream analysis."
查看一下结果
res
## 0 1 2 ## "Monocytes" "Neutrophils" "B cells"
添加到原来的对象中:
pbmc_small@meta.data$celltype <- as.factor(res[as.character(Idents(pbmc_small))])DimPlot(pbmc_small,group.by='celltype')

再对比一下手动注释的结果:

在大群的注释这一步几乎相同,唯一不足的是对于某一类亚群的注释不够准确,这也是原文中提到过的弊端(详见:)
06个性化修改:
如果想对某个细胞注释名进行修改也很简单:
res[1] <- 'Classical monocytes'pbmc_small@meta.data$celltype <- res[as.character(Idents(pbmc_small))]